Research
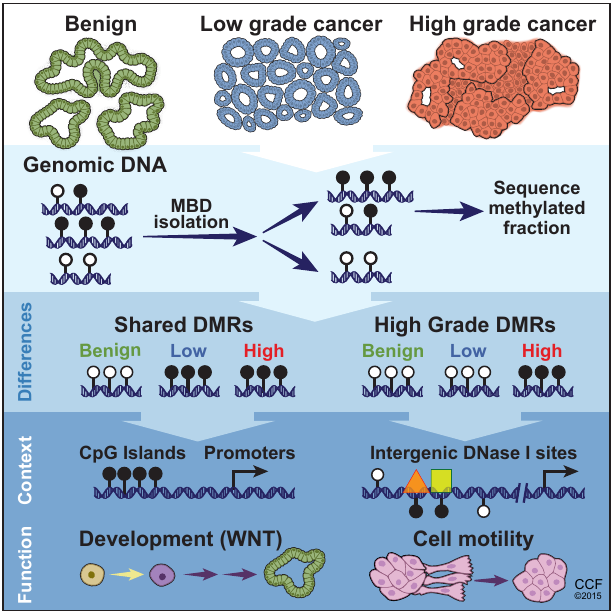
We found that gains of DNA methylation at certain loci can distinguish indolent from aggressive forms of prostate cancer. These genomic regions of focal hypermethylation fall in diverse genomic contexts, are enriched for regulatory elements, and correlate with the expression of genes linked to poorer outcomes. I created a web application (DMineR) allowing others to search and browse the complex set of epigenomic data generated by this work and integrate it with previous data from The Cancer Genome Atlas (TCGA). The bioinformatics demands of this study motivated the development of the Goldmine and MethylAction software, described below.
DMineR Web ApplicationRead in Cell Reports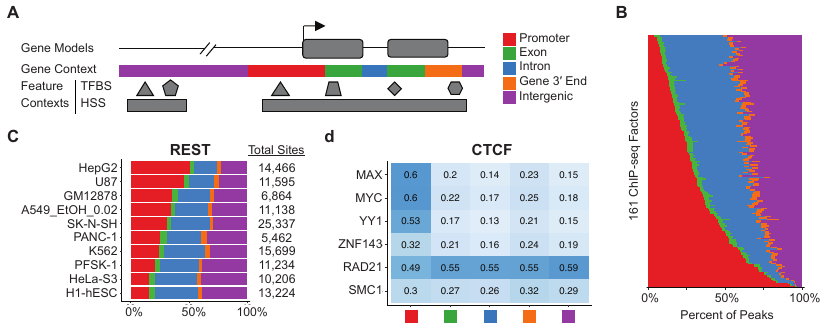
MethylAction and Goldmine are two R packages that meet separate analysis needs, but can also be used together, as they were in the prostate cancer and fibrosis studies also described on this page. MethylAction detects differentially methylated regions (DMRs) from the MBD-seq technique in comparisons involving any number of groups, and makes a substantial advance over existing tools. Goldmine takes a set of genomic regions, which could be DMRs from MethylAction or from any other source, and puts them into context with any number of reference data sets. This integration is invaluable for biologically interpreting the large sets of data generated by modern genomics.
Read MethylAction NAR PaperRead Goldmine NAR Paper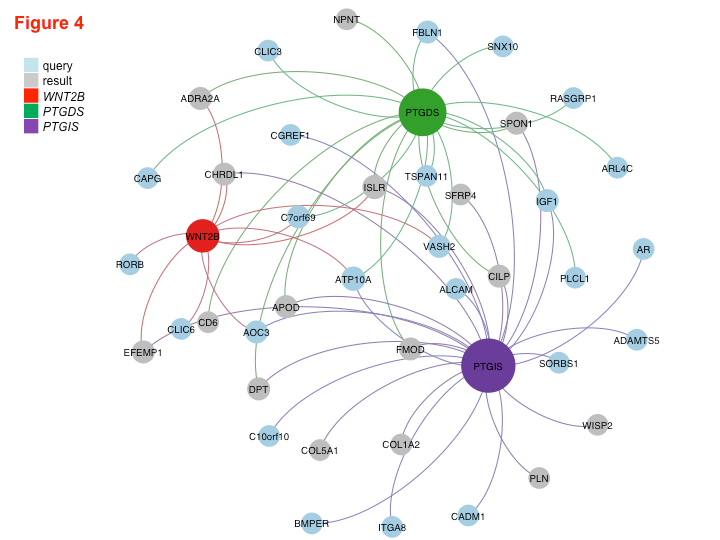
We analyzed both DNA methylation and RNA transcription genome-wide using MBD-seq and RNA-seq and integrated the two datasets to make discoveries about factors that drive fibrosis and determine if they could be regulated by DNA methylation. By better understanding the molecular networks that induce fibrosis, we aspired to identify new anti-fibrotic drug targets. This work, in collaboration with a pathobiologist, was an intriguing opportunity to apply my Goldmine tool to a new set of data outside of cancer biology.
Read in Clinical Epigenetics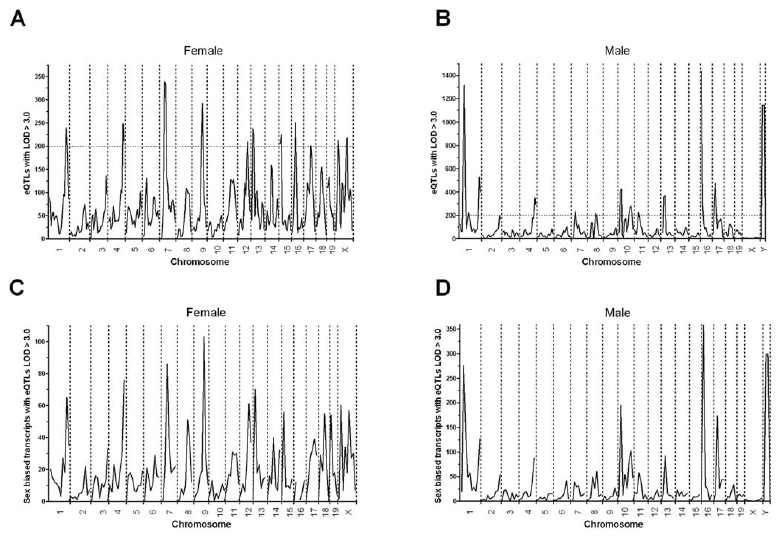
Right after my last day of high school classes, I immediately began working with Dr. Jonathan Smith at the Cleveland Clinic Lerner Research Institute. I performed QTL (quantitative trait loci) studies using the microarray technology available at the time, and we published two papers together about this work during my undergraduate career. The first discovered genetic associations with atherosclerosis, using lesion size as a quantitative trait and associating it with genotype genome-wide. Using my mastery of the bioinformatics tools involved, we further extended this work to perform an eQTL study, which associated gene expression with genotype transcriptome and genome wide. This work, published in my first lead-author paper, uncovered strong sex-specific effects of gene regulation.
Read in ATVBRead in PLoS ONE
Witnessing the publication of the human genome draft while in high school inspired me, at the encouragement of biology teacher Ann Brokaw, to seek out a university lab to learn genomics and bioinformatics. I started working with Dr. Evan Eichler and became fascinated by his work on the evolution of gene duplications using a technique called comparative genomics. When Dr. Eicher moved to the University of Washington, I began working with Dr. Mark Adams, who was the second author on Celera's publication of the human genome. I came up with the idea of uncovering the origin of known disease-causing mutations in humans. I hypothesized that the disease alleles in humans might actually be wild type (normal) in evolutionarily related species. After writing a complex software pipeline to analyze this question in detail, I found many examples that demonstrated my hypothesis, raising interesting avenues for future work in understanding the functional consequences of disease-causing mutations.
Coverage in USA TodayCoverage in NewsweekISEF Abstract & Photos